Moses Adondua Abah1,2 *, Esther Edesiri Ajoku3, Ekele Angel Ojimaojo1,2, Ekpenyong Utomobong Sunday4, Captain Tamunotonye Blessing5, Michael Abimbola Oladosu2,6 John Oladipo Olutayo7, and Nkwopara peace Chisa1
1Department of Biochemistry, Faculty of Biosciences, Federal University Wukari, Nigeria
2ResearchHub Nexus Institute, Nigeria
3Healthcare Management and Administration, University of New Orleans, 2000 Lakeshore Drive New Orleans, LA 70148, United States of America
4Department of Internal Medicine, College of Medicine, University of Uyo Teaching Hospital, Akwa Ibom State, Nigeria.
5Department of Plant Science and Biotechnology, Faculty of Science, University of Port-Harcourt, Nigeria
6Department of Biochemistry, Faculty of Basic Medical Sciences, University of Lagos, Lagos State, Nigeria
7Department of Chemical Sciences, Ramon Adedoyin College of Natural and Applied Sciences, Oduduwa University Ipetumodu, Osun State
*Correspondence: Moses Adondua Abah, Department of Biochemistry, Faculty of Biosciences, Federal University Wukari, Research Hub Nexus Institute, Nigeria E-mail: m.abah@fuwukari.edu.ng
Received: 18 Sep, 2025; Accepted: 06 Oct, 2025; Published: 15 Oct, 2025
Citation: Moses Adondua Abah, Esther Edesiri Ajoku, Ekele Angel Ojimaojo, Ekpenyong Utomobong Sunday, Captain Tamunotonye Blessing, Michael Abimbola Oladosu, John Oladipo Olutayo and Nkwopara peace Chisa. “Role of Inflammation in Atherosclerosis: Emerging Biomarkers and Therapeutic Targets” J Pathol Diagn Microbiol (2025). DOI: 10.59462/JPDM.1.1.102
Copyright: © 2025 Moses Adondua Abah. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Abstract
Atherosclerosis, a leading cause of cardiovas cular morbidity and mortality, is now recognized as a chronic inflammatory disease rather than a mere lipid storage disorder. Inflammatory pro cesses contribute to every stage of atheroscle rosis, from endothelial dysfunction and plaque formation to destabilization and rupture. Activat ed immune cells, cytokines, and adhesion mol ecules perpetuate vascular injury, driving dis ease progression. Recent advances highlight novel inflammatory biomarkers that improve risk prediction beyond traditional lipid profiles. Fur thermore, targeting inflammation offers promis ing therapeutic avenues for preventing adverse cardiovascular events. Understanding inflam mation’s central role is thus critical for identify ing emerging biomarkers and developing inno vative treatment strategies in atherosclerosis. The study highlights inflammation as a central mechanism in the initiation and progression of atherosclerosis. Findings reveal that pro-inflam matory cytokines, immune cell infiltration, and endothelial dysfunction collectively drive plaque development and instability. Emerging biomark ers such as high-sensitivity C-reactive protein, interleukins, and adhesion molecules demon strate strong predictive value for cardiovascular risk beyond traditional lipid measures. More over, therapeutic strategies targeting inflamma tion, including monoclonal antibodies and an ti-cytokine agents, show promising outcomes in reducing adverse cardiovascular events. These results emphasize the potential of integrating inflammatory biomarkers into clinical practice and advancing anti-inflammatory therapies as novel approaches to managing atherosclerotic cardiovascular disease. Inflammation is a pivot al driver of atherosclerosis, influencing plaque initiation, progression, and rupture. Emerging biomarkers provide valuable insights for early detection and risk stratification, while novel an ti-inflammatory therapies show potential in re ducing cardiovascular events. Integrating these advances into clinical practice could revolutionize prevention and treatment strategies. Contin ued research is essential to validate biomarkers and optimize therapeutic targets, ultimately im proving outcomes in patients with atherosclerotic cardiovascular disease.
Keywords:
Atherosclerosis, Inflammation, Biomarkers, Therapeutic targets, Immune cells, and Anti-inflammatory agents.
Introduction
Atherosclerosis is a chronic, progressive disease of the arterial wall characterized by the accumulation of lipids, fibrous elements, and immune cells, leading to the formation of plaques that can restrict or obstruct blood flow (Ayo et al., 2023). Traditionally, it was viewed primarily as a lipid storage disorder, where the deposition of cholesterol within the intima of arteries-initiated lesion formation. However, advances in molecular biology, vascular pathology, and immunology have transformed our understanding of this condition (Anih et al., 2025). It is now widely recognized that atherosclerosis is not merely a passive process of lipid accumulation, but rather a dynamic, inflammatory disease of the arterial wall. Inflammation plays a central role at every stage of atherogenesis, from the initiation of endothelial dysfunction and monocyte recruitment, to the progression of plaque instability and the eventual rupture that precipitates myocardial infarction or stroke (Anih et al., 2025). This evolving perspective has shifted research toward uncovering inflammatory biomarkers and developing therapies that target immune pathways in order to reduce cardiovascular risk beyond traditional lipid lowering strategies (David et al., 2025).
The inflammatory basis of atherosclerosis was first hinted at in pathological studies showing abundant immune cell infiltration within arterial plaques. Activated endothelial cells express adhesion molecules that promote monocyte and T-lymphocyte adhesion, while pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β) amplify the local immune response (Chen et al., 2016). Once within the intima, monocytes differentiate into macrophages, ingest modified low-density lipoprotein (LDL) particles, and form foam cells—hallmark features of early atherosclerotic lesions (Boch et al., 2021). Over time, these immune mechanisms contribute to plaque growth, necrotic core expansion, and extracellular matrix degradation, rendering plaques more vulnerable to rupture. Thus, the concept of atherosclerosis as a lipid-driven inflammatory disease has provided new insights into its pathophysiology and therapeutic opportunities (Bentzon et al., 2014).Importantly, inflammation in atherosclerosis is not confined to the arterial wall. It has systemic manifestations that can be detected through circulating biomarkers. High-sensitivity C-reactive protein (hs-CRP), one of the most studied inflammatory markers, has consistently been associated with increased cardiovascular risk, independent of lipid levels (Deguine and Barton, 2014). Similarly, novel biomarkers such as soluble adhesion molecules, chemokines, and cytokines have emerged as potential indicators of disease activity. The development of these biomarkers provides a window into the inflammatory milieu of the vasculature, offering clinicians tools for risk stratification, prognosis, and therapeutic monitoring (Citrin et al., 2021). The landmark JUPITER trial demonstrated that statin therapy not only reduced LDL cholesterol but also lowered hs-CRP, underscoring the dual lipid lowering and anti-inflammatory benefits of this drug class. More recently, the CANTOS trial provided direct evidence that targeting inflammation, specifically with the IL-1β inhibitor canakinumab—reduced cardiovascular events independent of lipid lowering (Chistiakev et al., 2017). These findings highlight the promise of integrating inflammatory biomarkers into clinical practice and the potential for anti-inflammatory agents as adjuncts in atherosclerosis management.
Despite these advances, significant challenges remain in translating inflammation-targeted therapies into widespread use. Inflammatory pathways are complex, redundant, and interconnected, raising concerns about immunosuppression, infection risk, and adverse systemic effects (Back et al., 2019). Moreover, not all inflammatory biomarkers are specific to atherosclerosis, as they may also reflect systemic inflammatory conditions such as infections or autoimmune diseases. Therefore, the identification of highly specific biomarkers and safe, effective therapeutic targets remains a priority in cardiovascular research (Badima and Vilahur, 2014). Advances in genomics, proteomics, and metabolomics are accelerating the discovery of novel mediators such as microRNAs, inflammasome components, and cell-derived extracellular vesicles, which may provide more precise insights into vascular inflammation (Bayer and Alcaide, 2021).
Emerging therapeutic targets extend beyond traditional lipid modifying and antihypertensive agents. Biologic therapies, such as monoclonal antibodies targeting cytokines, are being investigated for their potential to stabilize plaques and prevent recurrent cardiovascular events. Small molecule inhibitors of inflammatory signaling pathways, as well as therapies aimed at modulating immune cell phenotypes within plaques, represent promising strategies under investigation (Saad et al., 2025). Additionally, lifestyle interventions and nutraceuticals with anti inflammatory properties may complement pharmacologic therapies, reinforcing the concept of a holistic approach to vascular health. The recognition of inflammation as a pivotal driver of atherosclerosis represents a paradigm shift in cardiovascular medicine (Asada et al., 2020). No longer is the focus limited to lowering cholesterol or blood pressure alone; instead, it has expanded to encompass the regulation of immune responses that dictate plaque vulnerability and clinical outcomes (Moses et al., 2025). This broader perspective opens the door to personalized medicine, where the measurement of inflammatory biomarkers could guide individualized therapy, and novel anti-inflammatory agents could reduce residual cardiovascular risk that persists despite optimal lipid and blood pressure control (Ale et al., 2025).
The emergence of novel biomarkers provides opportunities for earlier detection, risk prediction, and therapeutic monitoring, while advances in immunomodulatory therapies promise to redefine treatment paradigms. Understanding the interplay between lipid metabolism and immune activation is therefore crucial for developing strategies that not only prolong life but also improve its quality by reducing the burden of atherosclerotic cardiovascular disease. As research continues to unravel the inflammatory underpinnings of this disease, the future of cardiovascular prevention and therapy may increasingly rely on targeting inflammation as a cornerstone of management. This review aimed at uncovering the role of inflammation in atherosclerosis with emphasis on the emerging biomarkers and therapeutic targets associated with this condition.
Prevalence of Atherosclerosis in Africa and the World at Large
In Nigeria, the situation reflects this global trend, but with unique national challenges. Historically, infectious diseases dominated Nigeria’s health landscape. However, in recent decades, rapid urbanization, lifestyle changes, and increased life expectancy have shifted the burden towards chronic non-communicable diseases, with atherosclerosis at the forefront (Ayo et al., 2023). Cardiovascular diseases are now among the leading causes of morbidity and mortality in the country. Research has shown that about one-quarter of Nigerian adults are at risk of developing coronary artery disease, a major complication of atherosclerosis. Hypertension, a primary risk factor, is highly prevalent, affecting roughly 31% of the adult population, and is frequently poorly controlled. Rising rates of diabetes, obesity, and dyslipidemia further compound the risk (Ayo et al., 2023; Saad et al., 2025).
Autopsy and clinical studies in Nigeria provide concrete evidence of the disease burden. For instance, an arterial autopsy study found that 20.8% of sampled individuals had advanced atherosclerotic lesions in their carotid arteries, indicating that significant vascular damage occurs silently before clinical symptoms appear (Jebari-Benslaiman et al., 2022). Similarly, hospital-based research has documented high rates of subclinical atherosclerosis among individuals with metabolic risk factors and people living with HIV on antiretroviral therapy. These findings highlight that, although national-level prevalence surveys are limited, atherosclerosis is already a significant health issue within the Nigerian population (Kleveland et al., 2018).
Atherosclerosis, the underlying cause of most cardiovascular diseases (CVDs), has become a major public health concern across the world. Globally, it accounts for the bulk of ischemic heart disease, ischemic stroke, and peripheral artery disease (Higashi et al., 2022). According to the Global Burden of Disease (GBD) study, ischemic heart disease alone affects more than 250 million people worldwide and causes nearly 9 million deaths annually, making it the single leading cause of mortality (He et al., 2022). While mortality rates have declined in many high-income countries due to advances in prevention and treatment, the absolute number of individuals living with atherosclerotic disease continues to rise, driven largely by population growth and aging. In lower- and middle-income regions, particularly Sub Saharan Africa, younger populations are increasingly showing signs of atherosclerotic disease, reflecting the global epidemiological shift towards non-communicable diseases (Guser and Sarapultsey, 2023).The global and Nigerian patterns underline the reality that atherosclerosis is no longer a disease of high-incomenations alone. The shift of its burden towards low- and middle-income countries, including Nigeria, underscores the urgent need for improved screening, early intervention, and health system preparedness. Without these, the nation may face an escalating crisis of cardiovascular disease in the coming decades.
Pathophysiology of Atherosclerosis
Atherosclerosis is a chronic, progressive disease of the arterial wall characterized by lipid accumulation, inflammation, and fibrosis that lead to narrowing and hardening of arteries (de Dekker et al., 2017). It underlies major cardiovascular disorders such as coronary artery disease, stroke, and peripheral arterial disease (Dutta et al., 2012). Its pathophysiology involves a complex interplay between endothelial dysfunction, lipid metabolism, inflammatory processes, and vascular remodeling.
The initiating event in atherosclerosis is endothelial injury or dysfunction, which may be caused by hypertension, hyperlipidemia, smoking, diabetes, or oxidative stress. Under normal conditions, endothelial cells regulate vascular tone, inhibit platelet aggregation, and maintain a non-thrombogenic surface (Koltsova et al., 2013). However, injury disrupts this balance, increasing endothelial permeability and expression of adhesion molecules such as VCAM-1 and ICAM-1. These changes promote the adhesion and migration of monocytes and T-lymphocytes into the intimal layer of the vessel wall (Lavin et al., 2020). A key step in early atherogenesis is the subendothelial retention of low-density lipoprotein (LDL) particles. LDL infiltrates the intima where it undergoes oxidative modification by reactive oxygen species. Oxidized LDL (oxLDL) is highly atherogenic, stimulating endothelial cells to release chemokines such as MCP-1, which attract more monocytes (Libby et al., 2010). These monocytes differentiate into macrophages, which engulf oxLDL via scavenger receptors, forming lipid-laden foam cells. The accumulation of foam cells produces the earliest visible lesion, known as the fatty streak.
As the lesion progresses, chronic inflammation becomes central to plaque development. Activated macrophages release pro-inflammatory cytokines (e.g., TNF-α, IL-1β) and proteolytic enzymes such as matrix metalloproteinases, which further damage the vascularwall. Smooth muscle cells (SMCs) from the media migrate into the intima in response to growth factors like platelet derived growth factor (PDGF) (Ayo et al., 2023). These SMCs proliferate and synthesize extracellular matrix proteins, including collagen and elastin, contributing to the formation of a fibrous cap over the lipid-rich necrotic core. This stage is known as the fibroatheroma. The stability of the atherosclerotic plaque depends largely on the integrity of the fibrous cap (Franks et al., 2023). Stable plaques are characterized by a thick cap and smaller lipid core, whereas vulnerable plaques have a thin cap, large necrotic core, and intense inflammatory activity, making them prone to rupture (Fenandez-Ruiz, 2016). Plaque rupture exposes thrombogenic material, such as tissue factor, to circulating blood, leading to platelet aggregation and thrombus formation. This can result in partial or complete arterial occlusion, causing acute clinical events such as myocardial infarction or ischemic stroke (Fallck-Hansen et al., 2013).
Other processes also contribute to plaque progression. Calcification occurs in advanced lesions due to osteogenic differentiation of vascular SMCs, further stiffening the arterial wall. Additionally, defective clearance of apoptotic cells (efferocytosis) leads to necrotic core expansion and increased plaque vulnerability (Gimbrane and GarciaCardenia, 2016). Neovascularization within the plaque may cause intraplaque hemorrhage, further destabilizing the lesion.
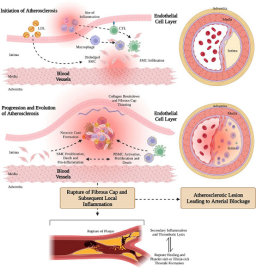
Figure 1. Atherosclerosis pathogenesis. From the illustration above, atherosclerosis pathogenesis begins with endothelial injury from factors like hypertension, smoking, or hyperlipidemia. This promotes LDL infiltration and oxidation, attracting monocytes that differentiate into macrophages. Foam cell formation, chronic inflammation, and smooth muscle proliferation lead to fatty streaks, fibrous plaque development, and eventual plaque rupture, causing thrombosis and vascular obstruction.
Source: Wolf and Ley (2019)
Inflammatory Mechanisms Underlying Atherosclerosis
Atherosclerosis involves lipid accumulation, immune activation, endothelial dysfunction, and maladaptive tissue remodeling. Inflammatory mechanisms underlie every stage of its development from initiation and plaque formation to progression, destabilization, and eventual clinical complications such as myocardial infarction, stroke, and peripheral arterial disease (Singh et al., 2023). Understanding these inflammatory processes is essential to identifying biomarkers and therapeutic targets for effective prevention and treatment.
Initiation: Endothelial Dysfunction and Immune Activation
The earliest step in atherosclerosis is endothelial dysfunction, often triggered by risk factors such as hypertension, hyperlipidemia, smoking, diabetes, and oxidative stress. Normally, the endothelium maintains vascular homeostasis by producing nitric oxide (NO), which regulates vasodilation and inhibits platelet adhesion (Singh et al., 2023). Under pathological conditions, oxidative stress reduces NO bioavailability and increases reactive oxygen species (ROS) production (Park et al., 2013). This leads to increased endothelial permeability, allowing low-density lipoprotein (LDL) particles to infiltrate the subendothelial space. Once trapped, LDL undergoes oxidative modifications (oxLDL), which are highly immunogenic and pro-inflammatory. Endothelial cells respond by upregulating adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), promoting the recruitment of circulating monocytes and T lymphocytes to the arterial wall. This endothelial activation marks the initiation of an inflammatory cascade (Nidorf et al., 2020).
Monocyte Recruitment and Differentiation
Circulating monocytes adhere to the activated endothelium and migrate into the intima under the influence of chemokines, especially monocyte chemoattractant protein-1 (MCP-1/CCL2). Within the intima, monocytes differentiate into macrophages, which express scavenger receptors such as CD36 and SR-A (Montarello et al., 2022). These receptors allow macrophages to internalize oxLDL particles in an unregulated manner, leading to lipid accumulation and the formation of foam cells. Foam cells represent the hallmark of early fatty streak lesions. Activated macrophages secrete pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), further amplifying local inflammation (Grindy et al., 2019). They also release proteolytic enzymes such as matrix metalloproteinases (MMPs), which degrade extracellular matrix (ECM) components and weaken the structural integrity of the arterial wall.
Role of Adaptive Immunity
Alongside innate immune responses, adaptive immunity plays a critical role in atherosclerosis. Antigen-presenting cells, including dendritic cells and macrophages, present oxLDL-derived antigens to naïve T cells, leading to their differentiation (Montarello et al., 2022). CD4+ T helper cells, particularly Th1 cells, release interferon-gamma (IFN-γ), a potent activator of macrophages that sustains inflammatory activity. Th17 cells, through secretion of IL 17, also contribute to vascular inflammation and plaque development (Singh et al., 2023). Conversely, regulatory T cells (Tregs) exert protective effects by secreting anti inflammatory cytokines such as IL-10 and transforming growth factor-beta (TGF-β), which help to dampen excessive immune activation (Park et al., 2013). The imbalance between pro-inflammatory Th1/Th17 responses and protective Treg activity is a critical determinant of plaque progression.
Progression: Chronic Inflammation and Necrotic Core Formation
As the lesion develops, chronic inflammation drives further tissue remodeling. Continued foam cell apoptosis and necrosis, coupled with impaired efferocytosis (clearance of dead cells), result in the accumulation of necrotic debris, forming a lipid-rich necrotic core (Fallck-Hansen et al., 2013). Necrotic core expansion promotes further release of danger-associated molecular patterns (DAMPs), which activate innate immune receptors such as Toll like receptors (TLRs) and NOD-like receptor protein 3 (NLRP3) inflammasome. Activation of the inflammasome leads to cleavage of pro-IL-1β and pro-IL-18 into their active forms, further fueling inflammation within the plaque (Fallck-Hansen et al., 2013).
Smooth muscle cells (SMCs), recruited from the media, migrate into the intima and produce extracellular matrix proteins such as collagen, forming a fibrous cap over the lesion. However, persistent inflammatory signals from macrophages and T cells weaken the fibrous cap by degrading collagen via MMPs and inhibiting collagen synthesis through IFN-γ. This dynamic interaction determines plaque stability (Park et al., 2013).
Plaque Destabilization and Rupture
The most dangerous clinical consequences of atherosclerosis occur when an inflamed plaque destabilizes and ruptures. Thin-capped fibroatheromas, characterized by large necrotic cores and thin fibrous caps, are highly vulnerable to rupture (Wolf and Ley, 2019). Inflammatory mediators such as TNF-α and IL-1β induce apoptosis of SMCs and promote further ECM degradation, weakening the cap. Upon rupture, the thrombogenic material within the necrotic core—such as tissue factor—comes into contact with circulating blood, triggering platelet aggregation and thrombus formation. This thrombotic event is the direct cause of acute coronary syndromes and ischemic strokes (Broch, et al., 2021).
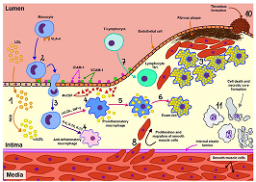
Figure 2. Summary of mechanisms underlying atherosclerosis. From the illustration above, the process begins with endothelial injury caused by factors such as hypertension, smoking, hyperlipidemia, or diabetes. This dysfunction promotes low density lipoprotein (LDL) infiltration into the intima, where it undergoes oxidation. Oxidized LDL triggers an inflammatory response, recruiting monocytes that differentiate into macrophages and engulf lipids, forming foam cells and fatty streaks. Smooth muscle cells migrate from the media to the intima, proliferate, and secrete extracellular matrix, leading to fibrous plaque development. Advanced lesions may undergo necrosis, calcification, and plaque instability, predisposing to rupture and thrombosis, which cause clinical events like myocardial infarction and stroke.
Source: Broch, et al. (2021)
Inflammatory Biomarkers in Atherosclerosis
Pentraxin
Pentraxin, known as C-reactive protein (CRP) is mostly produced in the liver in reaction to inflammatory cytokines, including TNF-alpha, IL-1, and IL-6 (Koltsova et al., 2013). Human coronary artery smooth muscle cells (HCASMCs) (Gimbrane and Garcia-Cardenia, 2016), human coronary artery endothelial cells (HCAECs) (Singh, et al., 2007) and sick coronary artery venous bypass grafts (Montarello et al., 2022). can also produce CRP in response to local inflammatory cytokines. The endoplasmic reticulum of cells first synthesizes it as monomers, which are subsequently put together to create pentamers (Gimbrane and Garcia Cardenia, 2016). Most research has concentrated on the well-established hsCRP as an inflammatory marker for ASCVD. HsCRP is used as a risk enhancer in the 2018 US multi-society guidelines for ASCVD risk assessment (Broch, et al., 2021). For instance, patients are deemed to be at higher risk if their hsCRP is greater than 2 mg/L and their 10- year risk score is intermediate (7.5–20%). The Physicians Health Study (Fenandez-Ruiz, 2016), one of the key preventative trials that supported this guideline, found that the highest quartile of hsCRP was linked to a 2.9-fold increased risk for MI and a 1.9-fold increased risk for stroke (p < 0.001, p = 0.02, respectively). Log-transformed hsCRP was then linked to a 1.23-fold higher risk for coronary heart disease, according to a meta-analysis of 54 prospective primary prevention cohorts conducted by the Emerging Risk Factors Collaboration (Koltsova et al., 2013). There were comparable results for ischemic stroke and vascular-associated risk (RR = 1.32; 95% CI = 1.18–1.49 and RR = 1.34; 95% CI = 1.20–1.50) (Gimbrane and Garcia-Cardenia, 2016). A meta-analysis of over 160,000 participants in a primary prevention study further supported these associations by finding an adjusted RR for coronary heart disease of 1.37 (95% CI 1.27–1.48), which is similar to more conventional risk factors like systolic blood pressure and non-high-density lipoprotein cholesterol (HDL-C). Even after controlling for conventional risk variables, these relationships were also highly predictive of ASCVD mortality (RR 1.55, 95% CI 1.37–1.76) and ischemic stroke (RR 1.27, 95% CI 1.15–1.40) (Koltsova et al., 2013). These results show that hsCRP is a significant and trustworthy risk marker for ischemic heart disease, but not a risk factor, since it is a surrogate marker of the interleukin (IL)-1β-to-IL-6-to-hsCRP pathway (Gimbrane and Garcia-Cardenia, 2016).
Interleukin-6 (IL-6) and Other Cytokines
Diffusible molecules with autocrine, paracrine, or endocrine activities are produced by interleukin-6 (IL-6), a type 2 cytokine that stimulates the humoral immune response. JAK/STAT, mitogen-activated protein (MAP) kinases, and Pi3k-Akt are examples of downstream transduction pathways that are triggered by IL-6 and IL-1 receptors (Koltsova et al., 2013). Though it comes from a variety of sites in the human body, macrophages, T helper cells, B cells, vascular endothelial cells, SM cells, and fibroblasts are the principal producers of IL-6 (Montarello et al., 2022). A causal association for the IL-6 pathway has been shown by Mendelian randomization studies that have shown a connection between genetic variation linked to plasma IL-6 levels and coronary events (Badima and Vilahur, 2014).
Lipoprotein-Associated Phospholipase A2 (Lp-PLA2)
Within atherosclerotic plaques, macrophages and macrophage-derived foam cells are the main producers of lipoprotein-associated phospholipase-A2 (LpPLA2), a plasma biomarker of inflammation. This enzyme is essential for the breakdown of oxidized phospholipids because it releases lysophosphatidylcholine and proinflammatory and pro-atherogenic oxidized fatty acids, which causes oxidative stress and accelerates the development of atherosclerotic plaque (Montarello et al.,2022). Lp-PLA2 is mostly linked to LDL, and research has indicated that elevated plasma levels, independent of non HDL-C levels, are linked to an increased risk of coronary artery disease (CAD) events. The Integrated Biomarker and Imaging Study 2 did not show any discernible cardiovascular benefits, even though darapladib, a specific Lp-PLA2 inhibitor, showed encouraging results in decreasing plaque volume (Badima and Vilahur, 2014). Therefore, without a successful targeted treatment with established ASCVD benefits, the usefulness of detecting Lp-LPA2 is still unknown.
Additional newly discovered indicators include myeloperoxidase, the link between MPO and CVD 1 has been explained by a number of molecular pathways (Gimbrane and Garcia-Cardenia, 2016). The NO system, endothelium, matrix proteinases, plasma lipoproteins, and atherosclerotic plaque are the most prevalent MPO targets associated with CVD. LDL is modified into chlorinated, nitrated, or carbamylated forms by MPO-generated species, mainly HOCl, HOSCN, and SCN−, which significantly increases the atherogenicity of LDL (Koltsova et al., 2013). Experimental research revealed that MPO products alter lipid and protein components. soluble ligand for CD40.
Atherosclerosis Treatment Targets
The Anti-Inflammatory Properties of Statins
Statins may lower inflammatory indicators, according to a number of clinical research. Pravastatin 40 mg daily, for instance, was reported to lower hsCRP levels longitudinally by 16.9% in the PRINCE study (Koltsova et al., 2013) and by 21.6% in a subset of patients from the CARE study (Montarello et al., 2022). It has also been demonstrated that the severity of statin medication contributes to the decrease in hsCRP. In the JUPITER study, for instance, individuals taking 20 mg of rosuvastatin saw a 37% decrease in their hsCRP levels (Gimbrane and Garcia-Cardenia, 2016). The reduction in CV risk was proportionate to the amount of hsCRP reduction attained in the JUPITER trial’s treatment group. Additionally, the primary endpoint was 55% lower for those who reached hsCRP levels of less than 2 mg/L than for those who did not (Fenandez-Ruiz, 2016). According to the PROVE-IT TIMI 22 research, 57.5% of patients who received treatment with atorvastatin 80 mg had a hsCRP of less than 2 mg/L (Fenandez-Ruiz, 2016). Furthermore, regardless of LDL-C, individuals who had hsCRP levels below 2 mg/L saw fewer CV events.
Anti-IL-1β Medications (e.g., canakinumab)
10,061 patients with a history of MI and hsCRP levels greater than 2.0 mg/L participated in the CANTOS phase III randomized placebo-controlled clinical trial, which showed the effectiveness of canakinumab, a monoclonal antibody (mAb) that targets IL-1β. When compared to a placebo, canakinumab at a dose of 150 mg resulted in a 15% decrease in the key composite endpoint of MI, stroke, and CV mortality, with an HR of 0.85 (95% CI 0.74–0.98) (Montarello et al., 2022). Important secondary outcomes were also reduced, such as MI, unstable angina requiring immediate revascularization, and any coronary revascularization. Nevertheless, there was no discernible decrease in either stroke or CV death. Notably, CANTOS demonstrated the clinical benefits of treating inflammation, marking a revolutionary moment. There was a decrease in cancer-related fatalities even though canakinumab treatment included a higher risk of deadly infection (Koltsova et al., 2013).
Colchicine
Colchicine, which is frequently taken for gout, familial Mediterranean fever, and off-label for pericarditis, suppresses cytoskeletal remodeling, phagocytosis, mitosis, and other intracellular activity. Among its many other functions, it also prevents inflammation caused by the NLRP3 infammasome (Fenandez-Ruiz, 2016). Colchicine was found to lower hsCRP levels by 60% in patients with stable CAD on statin therapy, according to small trials. The transcoronary gradients of IL-1β, IL-18, and IL-6 cytokines were significantly lower in patients with acute coronary syndrome (ACS) and stable CAD who were randomly assigned to receive colchicine (1 mg, followed by 0.5 mg 1 hour later) or no colchicine therapy prior to coronary angiography, according to other studies (Fenandez-Ruiz, 2016). The reductions ranged from 40 to 88%. The effectiveness of low-dose colchicine in lowering the incidence of recurrent ischemic cardiovascular events was more recently shown by the LODOCO2 (Montarello et al., 2022), and COLCOT (Gimbrane and Garcia-Cardenia, 2016). investigations. Over the course of 22.6 months, the COLCOT research, which included 4745 post-MI patients, showed a noteworthy 23% decrease in MACE, which includes CV mortality, cardiac arrest, MI, stroke, or urgent revascularization. The role of an anti-inflammatory drug in lowering the risk of MI is established by this trial. Following this, the LODOCO2 trial showed a 31% decrease in MACE when compared to a placebo, involving 2762 patients with chronic coronary disease who received modest doses of colchicine. hsCRP was not employed as an inclusion criterion in either experiment. In light of these findings, the FDA approved colchicine a broad label in 2023 to lower the risk of cardiovascular events in secondary prevention or in patients with many risk factors for cardiovascular disease (Koltsova et al., 2013).
Additional Newly Discovered Therapeutic Targets
In multiple clinical trials, tocilizumab, a humanized recombinant monoclonal antibody that targets the IL-6 receptor, has proven to be both safe and effective. It has been studied for secondary prevention of ASCVD and MI and has been licensed for the treatment of a number of autoimmune illnesses, including RA and giant cell arteritis (Broch, et al., 2021). A recent phase III ASSAIL-MI trial that was randomized, double-blind, and placebo-controlled (Broch, et al., 2021) revealed that the tocilizumab group had a higher myocardial salvage index than the placebo group. Additionally, there was reduced microvascular blockage in the tocilizumab arm; nevertheless, the size of the final infarct did not change significantly. 117 patients with non-ST segment elevation myocardial infarction (NSTEMI) participated in a recent double-blind, placebo-controlled experiment (Broch, et al., 2021). The tocilizumab group’s hsCRP levels were 2.1 times lower than the placebo group’s (2.0 vs. 4.2 mg/L/h, p<0.001) (Fallck-Hansen et al., 2013). Troponin levels were likewise decreased by tocilizumab (234 vs. 159 ng/ L/h, placebo versus tocilizumab, respectively, p=0.007), indicating that individuals receiving treatment experienced less myocardial damage than those receiving a placebo (Kleveland et al., 2018).
Challenges and Future Directions
Immunotherapy with a novel method shows promise for treating ASCVD. One of the most difficult issues is accurately detecting the clinical ASCVD phenotype and personalizing treatment based on individual patient characteristics such as immunological profile or genetic history (Broch, et al., 2021). Following the “lower the better” premise for LDL-C, the major purpose of ASCVD risk assessment is to identify individuals with high residual cholesterol. As a result, several lipid-lowering drugs have been developed, which, when taken with statin therapy, reduce the risk of ASCVD even further (Gimbrane and Garcia-Cardenia, 2016). Treatments that target residual inflammatory risk are based on the discovery of an inflammatory phenotype (most commonly measured by hsCRP) and information acquired over the last 30 years about the fundamental role inflammation plays in atherosclerosis (Koltsova et al., 2013). Furthermore, because the CANTOS study demonstrated the clinical significance of residual inflammatory risk, the role of inflammation in the development of ASCVD events is no longer a theory. Although hsCRP levels (a proxy marker of the IL-1β-to IL-6-tohsCRP pathway) are still used to identify residual inflammatory risk, therapy that lowers hsCRP to less than 2 mg/L produces RRR for MACE (Fig. 2). However, as shown in Figure 2, the extent to which each treatment reduces hsCRP and RRR varies. Regardless of LDL-C, it appears that a persistent inflammatory profile raises the risk of MACE (Fallck-Hansen et al., 2013). The findings that utilizing IL-6 as a measure of therapy response may help modify immunotherapy to reduce residual risk support the idea that additional biomarkers may aid in clinical decision-making. The varied disease states and inclusion criteria in therapeutic studies that target inflammation in ASCVD make adoption in a clinical setting difficult. Given the efficacy of immunotherapy in treating atherosclerosis, it is surprising that there are relatively few active trials in this area (Koltsova et al., 2013). Although imaging techniques Risk Populations in Africa.” (2025).can already detect inflammation, they are still rarely used in pharmaceutical research.
Conclusion
Inflammation plays a central role in the initiation, progression, and complications of atherosclerosis, making it a critical focus for both research and clinical management. Emerging biomarkers such as high-sensitivity C-reactive protein, interleukins, and adhesion molecules offer valuable insights for early detection and risk stratification. Additionally, novel therapeutic strategies targeting inflammatory pathways hold promise in complementing lipid-lowering therapies to reduce cardiovascular events. Continued exploration of inflammation-driven mechanisms and precision-based anti-inflammatory interventions will be vital for advancing personalized medicine, improving patient outcomes, and mitigating the global burden of atherosclerotic cardiovascular disease.
Acknowledgments
We want to thank all the researchers who contributed to the success of this research work.
Conflict of Interest
The authors declared that there are no conflicts of interest.
Funding
No funding was received for this research work.
References