Hasanin Mohamed Hasanin1*
M.B.B. Ch, M.Sc. Pediatrics, MD Pediatrics, Pediatric Department, Medical Research and Clinical Studies Institute National Research Centre
*Correspondence: Hasanin Mohamed Hasanin,M.B.B. Ch, M.Sc. Pediatrics, MD Pediatrics, Pediatric Department, Medical Research and Clinical Studies Institute National Research Centre.
E-mail: drhasanin1980@yah00.comReceived: 13 Feb 2026; Accepted: 10 Mar 2026; Published: 24 Mar 2026
Citation: Hasanin Mohamed Hasanin.“Updates on Management of Neonatal Cholestasis.” J Neonatol Pediat Care (2026):103.
DOI:10.59462/JNPC.2.1.103.
Copyright: © 2026 Hasanin Mohamed Hasanin.This is an open-ac cess article distributed under the terms of the Cre ative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Neonatal cholestasis (NC) is a life threatening, pathological condition that results from reduction or obstruction of the bile flow. It can affect all body systems and organs. So, early diagnosis is crucial to detect and refer patients to specialized medical centers to improve their management and prognosis [1,2] NC should be suspected when prolonged jaundice occurs in formula fed infants who remain jaundiced at 2 weeks of age and breastfed ones who remain jaundiced at 3-4 weeks of age [3] In the past, NC can be defined as conjugated bilirubin >1 mg/dL (if total <5 mg/dL) or>20% of total serum bilirubin (if total >5 mg/dL) with passage of dark colored urine staining the diapers with or without acholic stools. NC is never physiologic and should always promptly evaluated to determine the specific etiology. Certain etiologies, including galactosemia, tyrosinemia, choledochal cyst, bacterial and viral sepsis, and biliary atresia (BA), are amenable to medical or surgical interventions that may prevent progression to liver failure or the development of serious extrahepatic manifestations. Regardless of etiology, infants with cholestasis benefit from optimizing nutrition with fat soluble vitamin supplementation and preventing complications of liver disease. including cholangitis, pruritus, portal hypertension, and ascites [4,5].
Recently, according to North American Society of Pediatric Gastroenterology, Hepatology, Nutrition and European Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN, ESPGHAN Guidelines 2022). NC can be defined as conjugated (direct) bilirubin > 1 mg/dL (17.1 mcmol/L) in an infant regardless of the total bilirubin level or conjugated bilirubin > 20% of total when total serum bilirubin level > 5 mg/dL (85.5 mcmol/L). In practice, lower thresholds are used for considering a diagnosis of cholestasis in the first 5 days of life including conjugated bilirubin level meeting any of the following:
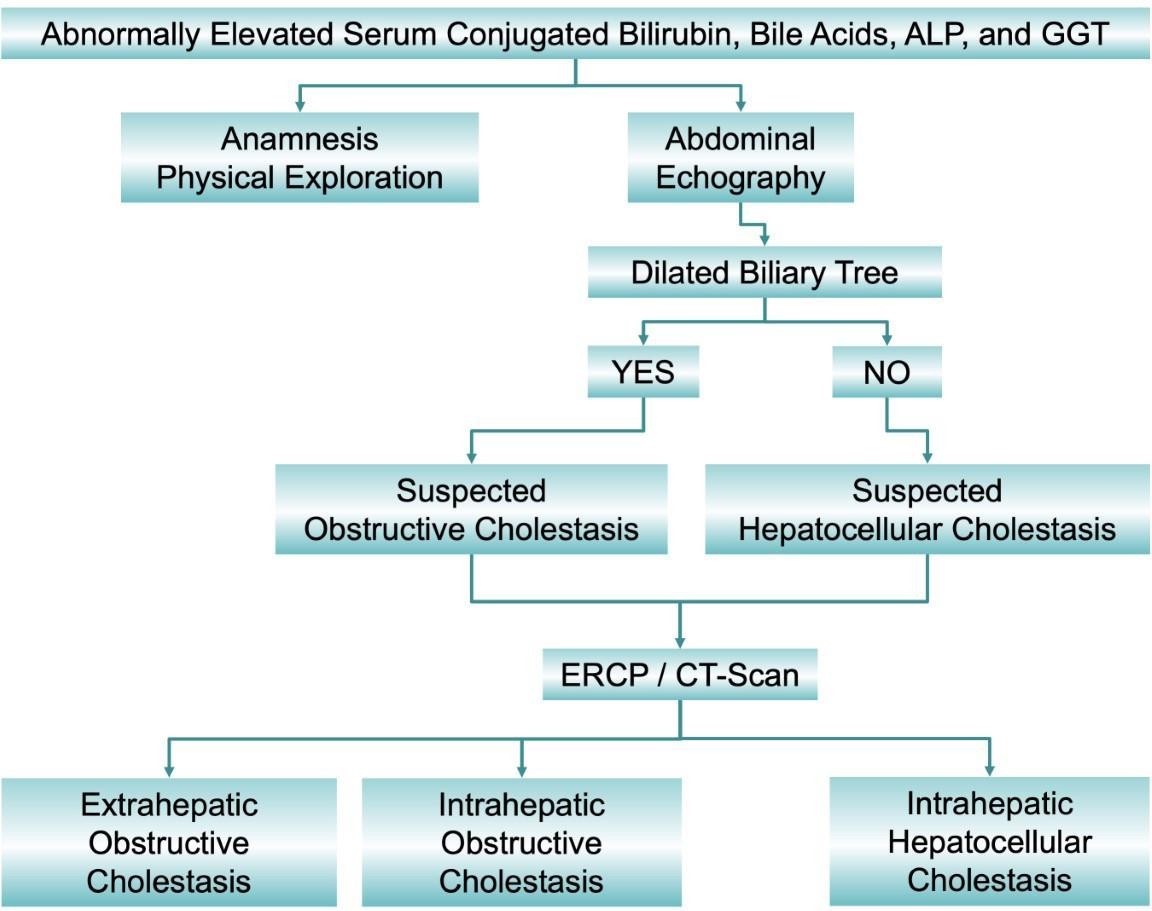
Figure 1.Classification of cholestasis according to biochemical data, image information obtained by abdominal echography, endoscopic retrograde cholangiopancreatography (ERCP) and computed tomography scan (CT-Scan), and the anatomical location of the cause. ALP: alkaline phosphatase; GGT: γ- glutamyle transferase.[6]
| Anatomic obstruction |
|---|
| Biliary atresia, choledochal cyst, cholelithiasis, biliary sludge, inspissated bile syndrome and bile duct tumors. |
| Infections |
| Viral, bacterial, parasitic, spirochete |
| Toxins |
| Drugs as ceftriaxone, erythromycin, rifampicin, TPN |
| Endocrine |
| Hypothyroidism- panhypopituitarism |
| Immune |
| Gestational alloimmune liver disease (neonatal hemochromatosis previously) |
| Genetic and inborn errors of metabolism |
| ἀ1-antitrypsin deficiency (A1AT) |
| Alagille syndrome (ALGS) |
| Arthrogryposis-renal dysfunction-cholestasis (ARC syndrome) |
| Congenital hepatic fibrosis |
| Citrin deficiency |
| Cystic fibrosis (CF) |
| Bile acid synthesis defect |
| Bile acid conjugation defect |
| Fatty acid oxidation defect |
| Galactosemia |
| Tyrosinemia |
| Glycogen storage disease type IV |
| Mitochondrial respiratory chain disorders |
| Neonatal ichthyosis-sclerosing cholangitis |
| Neonatal sclerosing cholangitis |
| Niemann pick type C disease |
Table 1.Causes of Neonatal Cholestasis.
The overall incidence of neonatal cholestasis is approximately 1 in 2,500 live births. For extremely premature babies
it is much higher, due to the combination of several risk factors such as immaturity, lack of enteral feedings, long
term use of total parenteral nutrition, and frequent episodes of septicemia. [7]. Cholestatic jaundice can result
from various causes, including infections, anatomic abnormalities of the biliary system, endocrinopathies, genetic
disorders, metabolic abnormalities, toxin and drug exposures, parenteral nutrition (PN) administration, cardio
vascular dysfunction, and neoplastic processes [8]
The most common identifiable causes include BA (25–40%), genetic and metabolic diseases, including α1 antitrypsin
(A1AT) deficiency (10–20%), Alagille syndrome (ALGS 2–14%), cystic fibrosis (CF), progressive familial intrahepatic
cholestasis (PFIC), hypopituitarism (5%), inspissated bile syndrome, idiopathic neonatal hepatitis (INH) or
transient neonatal cholestasis (TNC); and PN associated cholestasis (PNAC) in preterm infants and those with
intestinal failure. All the previous causes can be summarized in table 1 below [9]
According to a meta-analysis, published in 2015 and analyzed data of 1,692 cases of NC from 17 studies, biliary
atresia (BA) was the most common cause worldwide (26%); other etiologies include infection (12%), total parenteral
nutrition (6%), metabolic disease (4.3%), alpha-1 antitrypsin deficiency (4.1%), and perinatal hypoxia/ischemia
(3.6%). Much of the literature to date originated from Caucasian and Asian populations, thus the epidemiology and
pattern of NC remain unclear among Arabs. This missing information would have a great impact on the diagnostic
algorithm used and the choice of laboratory tests in real clinical practice as shown in figure 2 below.
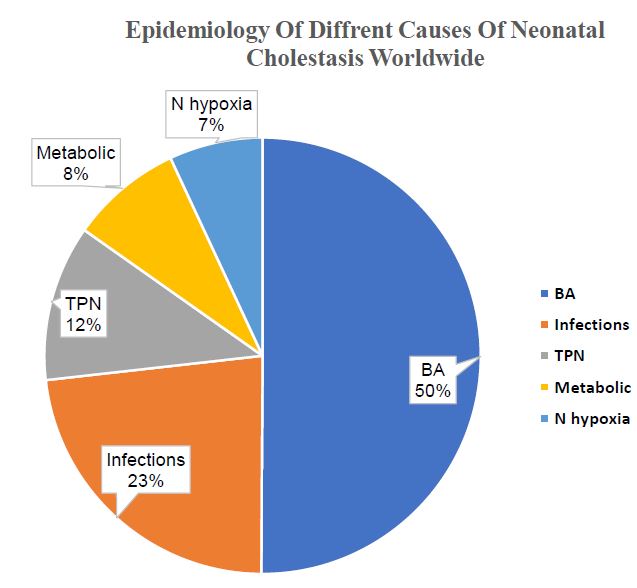
Figure 2:Epidemiology of Neonatal Cholestasis worldwide. [10]
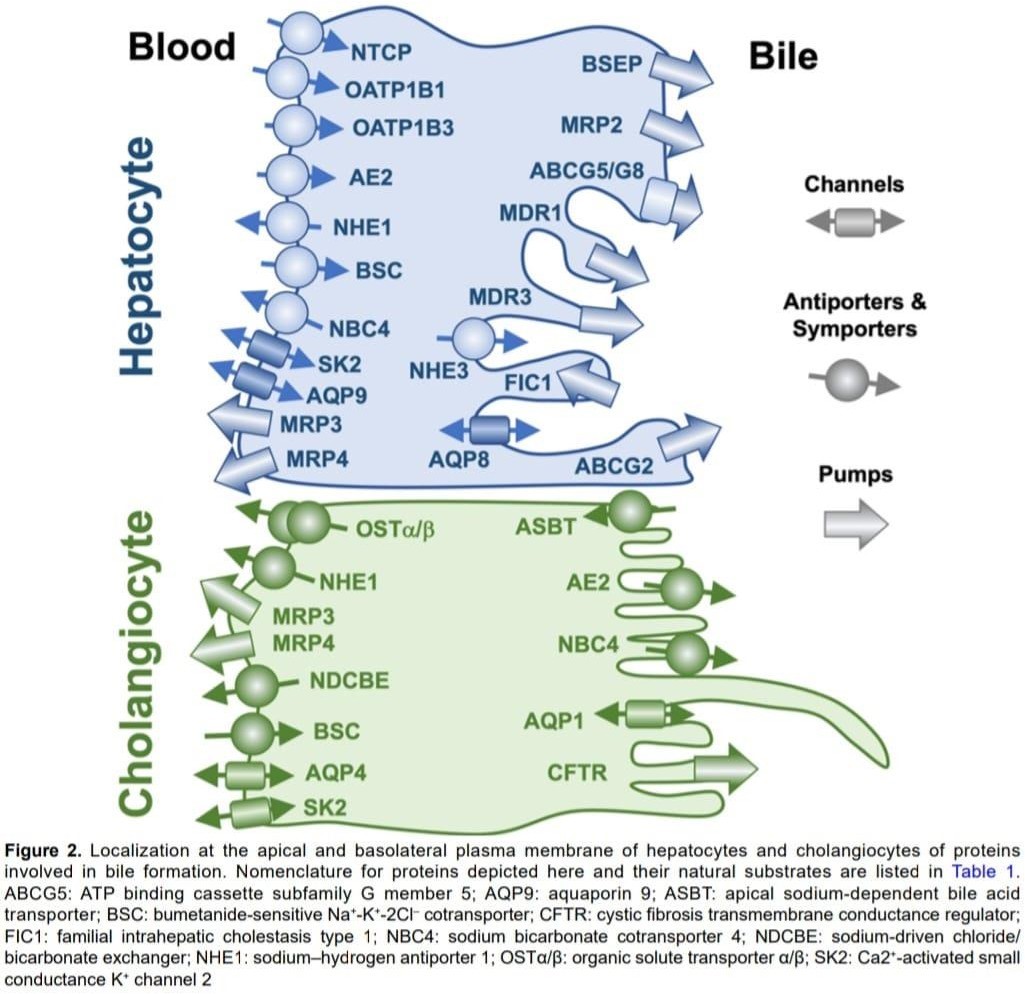
Cholestasis occurs when bile flow is impaired, leading to accumulation of bile acids in the liver and bloodstream as mentioned in figure 3 below [11]. Key pathophysiological mechanisms:
Hepatic Level
Extrahepatic Level
Molecular Mechanisms
Infants with cholestasis often have olive green jaundice indistinguishable from that of infants with indirect hyperbilirubinemia; however, the presence of pale stools, dark yellow urine, or hepatomegaly should suggest cholestasis. Moreover, one must not be falsely reassured by improving jaundice over the first few weeks after birth, as this can be the result of an improving indirect bilirubin portion (from physiologic jaundice, breast milk associated jaundice, or a resolving cephalohematoma), while the direct portion remains high or even rises. Acholic stools signify biliary obstruction and should always prompt further evaluation [12].
Specific clinical findings that suggest cholestatic disorders include bleeding or bruising results from vitamin K induced coagulopathy (ie, vitamin K malabsorption in A1AT deficiency or ALGS) or from liver failure (eg, in gestational alloimmune liver disease, tyrosinemia, or mitochondrial respiratory chain disorders) [13]. Splenomegaly can result from cirrhosis with associated portal hypertension, storage diseases, hemolytic disorders, or congenital hemophagocytic lympho-histiocytosis. Neurologic abnormalities, including irritability, lethargy, poor feeding, hypotonia, or seizures, can be found in infants with infection, intracranial hemorrhage, metabolic (eg, Zellweger syndrome) and mitochondrial disorders, or hyperammonemia from liver failure. Infants with congenital infection may have low birthweight, thrombocytopenia, petechiae and purpura, and chorioretinitis. Facial dysmorphisms can be observed in chromosomal disorders or ALGS [14]. Cardiac murmurs, particularly those associated with the pulmonary artery, are frequent in patients with ALGS or BA. It is critical to keep in mind that many infants with BA will appear healthy during the first month of age, and hepatosplenomegaly, poor growth, and even acholic stools may escape early identification [15].
Biliary atresia
BA is the most common cause of liver transplantation in pediatric patients and needs urgent management to prevent liver cirrhosis. BA is a unique disease to the neonatal period representing the end result of a damaging inflammatory process with unclear a etiology affecting both intra and extrahepatic bile ducts [16]. Four different variants of BA can be distinguished based on clinical or laboratory features, isolated disease, cystic BA, virally associated BA especially with cytomegalovirus infection and BA with splenic malformation syndrome. NC is the key feature of BA especially when associated with pale stool and dark urine in a healthy infant. The biochemical features of the disease include direct hyper bilirubinemia, raised liver transaminases, raised alkaline phosphatase and γ-glutamyl transpeptidase however, these findings may overlap with many other causes of NC. Non-dilatation of biliary tract, absent or non-contractile gallbladder, positive triangular cord and sub capsular hepatic flow, and right hepatic hypertrophy are the main findings by abdominal ultrasound that help in the diagnosis of BA. The presence of bile duct proliferation, bile plug, a small cell infiltrate, and portal fibrosis and the absence of sinusoidal fibrosis and giant cells are the major histopathological findings in BA. [17]. Duodenal aspiration and analysis for bile have been used for diagnosis of BA. Because of the poor differentiating rule of technetium labelled iminodiacetic acid derivatives, this technique now is less commonly used. Endoscopic retrograde cholangiopancreatography is another recent tool for diagnosis of BA, but it highly invasive. On table cholangiography remains the gold standard for diagnosis [18].
The usual management of BA is a surgical attempt to restore bile flow using the Kasai portoenterostomy technique. Recent studies on BA have been focused on non- invasive methods to predict liver fibrosis progression A 2023 study published in Frontiers in pediatrics explored the predictive value of APRI (aspartate aminotransferase to platelet ratio index) and FIB 4(four factor-based fibrosis index) score in diagnosing progressive liver fibrosis in children with BA. APRI had 52% sensitivity and 83% specificity while FIB4 had 83% sensitivity and 67% specificity. These markers can be used as follow up for progressive fibrosis in BA patients.
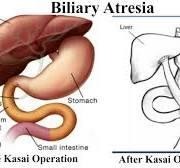
Figure 4:Kasai operation for biliary atresia (BA). [19].
It is a congenital dilation of the biliary tree not secondary to an obstruction. It is a benign condition but can be complicated by cholestasis, cholelithiasis, cholangitis, biliary cirrhosis, pancreatitis, and malignant transformation [20]. Abdominal ultrasound demonstrates abnormal dilatation of the CBD. Laboratory results reveal conjugated hyperbilirubinemia, increased γ-glutamyl transpeptidase (GGT) level, and mild elevation of liver transaminase levels. Albumin and globulin levels are normal. It is essential to differentiate it from cystic BA; Surgical cyst excision is the treatment of choice. Laparoscopic hepaticojejunostomy is a new feasible treatment option achieving better outcome than traditional treatment modalities with fewer complications. [21].
IBS is a rare cause of NC resulting from the obstruction of the EHBDs by either bile plugs or sludge without chemical defects of the bile, anatomical abnormalities, or liver cell damage. IBS accounts for 8% of the surgically treatable causes of NC, with an estimated incidence of 1/175,000 live births [22]. Systemic infection, hemolysis, total parental nutrition (TPN), rapid weight loss, progressive familial intrahepatic cholestasis (PFIC), citrine deficiency, and drugs (ceftriaxone) can cause IBS. IBS can resolve spontaneously with or without the administration of oral ursodeoxycholic acid (UDCA) (10–20 mg/kg). Failed medical management indicates the need for endoscopic retrograde cholangiopancreatography, percutaneous transhepatic cholangiography, or irrigation of the biliary tree with saline or a mucolytic agent through a cholecystostomy. Co- administration of N-acetylcysteine and glucagon can effectively treat IBS. Omega- three polyunsaturated fatty acids (500 mg four times per day) can be used as an alternative to surgical intervention [23].
It is a rare condition that affects children under four years old, with most cases reported in six-month-old infants [1]. The exact cause of SPBD is still unclear, but research suggests it may be linked to pancreaticobiliary malunion (PBM) and congenital weakness of the bile duct wall. Clinically, it can present with fever, nausea and vomiting, abdominal pain and distension. Diagnosing (SPBD) can be challenging due to its non-specific clinical manifestations. Laboratory tests, imaging modalities such as abdominal CT scan and paracentesis are essential for diagnosis. Treatment is either conservative medical or surgical (Roux-en-Y anastomosis) for severe cases [24].
Idiopathic neonatal hepatitis (INH) is a condition characterized by inflammation of the liver in newborns, with unknown causes. It's also known as idiopathic neonatal cholestasis, it is usually presented with prolonged conjugated jaundice, poor weight gain (failure to thrive), fat soluble vitamins deficiency and pale or clay colored stool. It is diagnosed after exclusion of other causes of cholestasis such as BA, choledochal cyst, alpha-1-antitrypsin deficiency, Alagille syndrome, Progressive familial intrahepatic cholestasis (PFIC), Congenital infections and Cystic fibrosis [25].
Sepsis Inflammatory mediators (i.e., bacterial endotoxin and lipopolysaccharides) cause NC by triggering the release of cytokines. NC is considered to be a severe condition that may complicate neonatal septicemia. Prolonged NC will aggravate liver dysfunction, leading to liver failure and the failure of other organs. Management of neonatal sepsis-associated NC at the early stage is mandatory to prevent its sequelae and maintain normal growth and development [26].
ἀ1 antitrypsin deficiency (A1ATD)
Alpha-1 antitrypsin deficiency (AATD) is a genetic disorder affecting the production of alpha-1 antitrypsin, a protein protecting lungs and liver from damage. It is usually presented with pulmonary manifestation such as chronic obstructive pulmonary disease (COPD), emphysema and bronchiectasis. Hepatic manifestations such as chronic liver disease, hepatitis and cholestasis. Diagnosis includes AAT measurement, pulmonary functions test and genetic testing for SERPINA 1 gene [27].
Galactosemia
Galactosemia is a rare genetic metabolic disorder affecting the body's ability to metabolize galactose, presented with jaundice, vomiting, failure to thrive, diarrhea. Developmental delay (speech language and cognitive function). Recently, its diagnosis depends mainly on neonatal screening program to detect enzyme deficiency early. In addition to, urinary galactose test and genetic testing. Treatment is galactose free diet and avoidance of dairy products and use of soy-based or elemental formula [28].
Tyrosinemia
Tyrosinemia is a rare genetic disorder affecting tyrosine metabolism.
Types
Clinically, it can be presented with hepatic dysfunction (cholestasis, liver cirrhosis, hepatocellular carcinoma), renal manifestation (Fanconi syndrome, aminoaciduria), neurological (seizures and intellectual disabilities) and ocular manifestations (corneal ulcers, glaucoma). Diagnosis occurs by early neonatal screening (blood test for tyrosine and succinyl acetone), biochemical tests (urinary succinyl acetone and plasma amino acids) and recently genetic testing for mutated enzymes. It should be diagnosed as early as possible due to possibility of hepatocellular carcinoma (Masurel-Paulet, 2023).
Progressive familial intrahepatic cholestasis is a rare genetic disorder characterized by impaired bile flow and accumulation of bile acids in the liver It's classified into three main types:
Types of PFIC
PFIC Type 3 (MDR3 deficiency): Caused by mutations in the ABCB4 gene, leading to mild to moderate liver disease with variable progression [30]. Clinically, PFIC can be presented with pruritus, jaundice, and failure to thrive. Laboratory tests reveal elevated bile acids, low gamma glutamyl transferase (GGT), and abnormal liver function tests. Diagnosis is confirmed by genetic testing and liver biopsy. Treatment is supportive to prevent liver damage, ursodeoxycholic acid (UDCA) for pruritis, biliary diversion and finally liver transplantation is the last option for severe refractory cases [31].
Genetic Disease
Cystic fibrosis is a genetic disorder that affects the respiratory, digestive, and reproductive systems. It's characterized by thick, sticky mucus that obstructs the alveoli of the lungs and the pancreas [32]. CF is caused by mutations in the CFTR gene, with the p. Phe508del mutation being the most common. It affects approximately 1 in 50,000 to 100,000 births. Clinically, it can be presented with persistent cough, wheezing, poor growth or weight gain, and frequent respiratory infections. Gastro-intestinal manifestations (recurrent vomiting, meconium plug syndrome, steatorrhea due to pancreatic insufficiency, malabsorption of fat-soluble vitamins (A, D, E, K) and cholestasis. Diagnosis is typically made through sweat chloride test, genetic testing, and newborn screening. Treatment options include CFTR modulators, which target the defective protein, antibiotic therapy, nutritional support, and lung transplantation in severe cases. Recent studies have focused on understanding the mutations and their effects on CFTR protein function, leading to personalized medicine approaches. The introduction of CFTR modulators has been a significant advancement in CF therapy, offering improved lung function and quality of life for many patients [33].
Citrin deficiency
Citrin deficiency is a rare genetic disorder caused by mutations in the SLC25A13 gene, leading to impaired citrin protein function. Citrin plays a crucial role in mitochondrial aspartate-glutamate transport. [34]. Clinically, presented with early cholestasis and liver dysfunction, poor weight gain (failure to thrive), developmental delay and specific laboratory parameters in the form of hypoglycemia, increased ammonia level and lactic acidosis. Diagnosis is made by Plasma amino acid, urine organic acid analysis and newborn screening (blood test for citrulline and arginine). Recently, genetic testing via SLC25A13 gene sequencing and liver biopsy. Treatment includes low protein diet, arginine supplementation to reduce ammonia level and finally liver transplantation is the last option in severe cases [35].
Alagille Syndrome
Alagille syndrome (ALGS) is a rare genetic disorder affecting multiple systems, primarily the liver, heart, skeleton, face, and eyes. It is characterized by AD inheritance. Its prevalence is reported to be approximately 1 in 30,000 infants (Kamath, 2022). Clinically, presented with cholestatic jaundice, facial features (triangular face, prominent forehead, deep set eyes, pointed chin, and straight nose), cardiac defects (peripheral pulmonary artery stenosis), skeletal abnormalities (butterfly vertebrae), ophthalmological (posterior embryotoxic) and renal defects.
According to the Guidelines of the American Association for Study of Liver Diseases 2022[36], diagnosis is mainly through genetic testing by identifying mutations or deletions in the JAG1 gene on chromosome 20p11.2-20p12, responsible for approximately 98% of cases. Treatment is mainly supportive until end stage liver disease occurs at which liver transplantation is mandatory.
BASDs are disorders of primary bile acid (cholic and chenodeoxycholic acid) synthesis that result in liver injury due to the accumulated toxic intermediate metabolites. Abnormalities in bile excretion result in the retention of other toxic metabolites within the liver. BASDs should be included in the differential diagnosis of NC [37]. The effects of BASDs on the liver range from persistent cholestasis to acute hepatitis or liver failure.
Clinically, patients present with NC, FTT, hepatosplenomegaly, rickets, evidence of fat malabsorption, and bleeding. Neurologic effects include seizures, developmental delay, deafness, blindness, and neuromuscular weakness [38]. UDCA disrupts serum bile acid levels, and liver transaminases are elevated with normal GGT. Urinary bile acids should be measured to identify the synthetic defect. Liver biopsy is not diagnostic. Treatment with cholic acid, not ursodiol, suppresses the production of toxic metabolites and maintains normal growth and development
NC is a frequently reported complication of TPN. TPN-AC is diagnosed if there is persistent conjugated hyperbilirubinemia greater than 2 mg/dL for at least two consecutive tests during TPN, with the absence of any other causes of NC [39]. The exact aetiology is not known; risk factors associated with TPN-related cholestasis are very low birth weight, prematurity, the duration of TPN, sepsis, the absence of enteral feeding, the quality or quantity of amino acid intake, male sex, trace mineral or phytosterol toxicity, and perinatal depression or shock. Intestinal resection and its complications have also been associated with the occurrence of TPN-AC. The severity of the disease varies from mild to severe, and it can lead to significant hepatic injury and end stage liver disease. The histopathological changes are correlated with the duration of TPN. A shortened TPN course and early initiation of enteral feeding can effectively decrease the frequency of NC. UDCA (10–30 mg/kg/day) is the most widely used drug in the treatment of TPN-AC [40].
Diagnostic Work UP
If cholestasis is present, further evaluation should be completed with a sense of urgency to identify or exclude BA and other treatable conditions (e.g., hypothyroidism, urinary tract infection, sepsis, galactosemia, and tyrosinemia). A careful family history and physical examination should be performed that might point to a specific etiology. All infants with cholestasis should initially undergo testing for A1AT deficiency which is a common cause of cholestasis with an A1AT serum level and/or electrophoresis/genotype testing (a level of <50 mg/dL [9.2 µmol/L] or a ZZ/SZ genotype suggests disease). The newborn screen should be reviewed or specific testing ordered to evaluate for hypothyroidism, tyrosinemia, or galactosemia, which are all treatable conditions. If the infant has a fever, abnormal urine analysis findings, or other clinical signs of infection, appropriate blood, urine, and/or spinal fluid cultures should be performed. Abdominal ultrasonography can identify physical obstruction of the biliary tree (ie, a gallstone, choledochal cyst, or mass) or gall bladder abnormalities associated with BA. If these tests do not reveal an etiology for the infant’s cholestasis, and if a liver biopsy can be performed safely, biopsy should be performed to evaluate for BA. If red flags for individual diseases are present then specific diagnostic tests should be done [41] as mentioned in figure 3 below. In the past, multiple individual blood and urine tests were used simultaneously to evaluate for various genetic/metabolic causes of cholestasis after A1AT deficiency and BA were excluded; these tests include.
Unfortunately, this single test approach was timely and expensive. So, the paradigm for evaluating cholestasis has now changed at many institutions where next generation sequencing (NGS) targeted genetic panels, whole exome sequencing (WES), and/or whole genome sequencing (WGS) are clinically available in a relatively rapid, cost-effective manner. This testing can identify all known gene mutations associated with cholestatic diseases and also uncover new genetic abnormalities resulting in cholestasis. (Karpen et al., 2017).
Imaging
All infants with cholestasis should undergo abdominal ultrasonography as part of their initial evaluation to assess liver structure, size, intra-abdominal position, composition and to identify number of spleens (asplenia or polysplenia can occur in BA, splenic malformation syndrome); evaluate for the presence of ascites; and identify findings of an extrahepatic obstructive lesion (choledochal cyst, mass, gallstone, obstructing sludge). Although ultrasound findings such as the absence of the gallbladder, presence of a triangular cord sign (a cone-shaped fibrotic mass cranial to the bifurcation of the portal vein), interrupted inferior vena cava, preduodenal portal vein, situs inversus, and heterotaxia, may suggest BA, none of these findings are diagnostic for BA, and conversely, some infants with BA have normal hepatobiliary ultrasonography. [22].
In the past, hepatobiliary scintigraphy with a technetium labeled iminodiacetic acid analogue (HIDA) was used to assess biliary patency and rule out BA. However, its specificity is low (33%–80%); non-excretion by a HIDA scan is observed not only in infants with BA, but also in those with intrahepatic cholestasis, including infants with ALGS and INH, or those receiving PN. (Nievlstein et al., 2021). It is recommend reserving the HIDA scan for preterm infants with cholestasis in whom the suspicion of BA is low (ie, an infant with pigmented stool who is receiving PN) and in those for whom liver biopsy would be high risk. (Kiamifar et al., 2013). Cholangiography is ultimately required to establish the diagnosis of BA, and this is most commonly performed as an intraoperative cholangiography (IOC) by a surgeon experienced in performing HPE during the same surgical procedure if the IOC shows BA. At this time, endoscopic retrograde cholangiopancreatography, magnetic resonance cholangiopancreatography, and transhepatic cholecystography are of limited usefulness in the evaluation of neonatal cholestasis, except in specialized centers [43].
Liver Histology
Percutaneous liver biopsy remains a critical tool in evaluating neonatal cholestasis. Several studies have found that a diagnosis of BA was correctly suggested by liver biopsy histology in 85% to 95% of cases (including preterm and term infants). The histologic features that distinguish cases of BA from other causes include:
Also, liver biopsy is valuable in diagnosis of other causes of NC such as:
Although liver biopsy is the investigation of choice in diagnosis of NC, it is an invasive maneuver that many complications can result from it. In BA, hepatic fibrosis is a universal feature, and biliary cirrhosis occurs early in life, its precise timing varies depending on the child clinical condition. Recently, noninvasive methods to evaluate liver stiffness and fibrosis (ie, acoustic radiation force impulse, transient elastography, or sheer wave elastography) could be used to accelerate suspicion of BA and shorten the time to IOC and HPE [45].
Recent studies have suggested that elevated serum levels of matrix metalloproteinase 7 (MMP7) at 1 to 2 months of age
may reliably discriminate BA from other causes of cholestasis, with positive predictive values exceeding 90% and
negative predictive values exceeding 95% [46].
Further studies are needed to determine the earliest age at which
MMP7 can be used, to clarify whether MMP7 is reliable in unique populations of cholestatic infants, including
preterm infants and infants with congenital heart disease, and finally, to assess whether MMP7 levels can
prognosticate which BA infants will ultimately need liver transplantation [47].
The algorithm for evaluation of
neonatal cholestasis (including different causes, their laboratory investigations) can be summarized in figure 5
below.
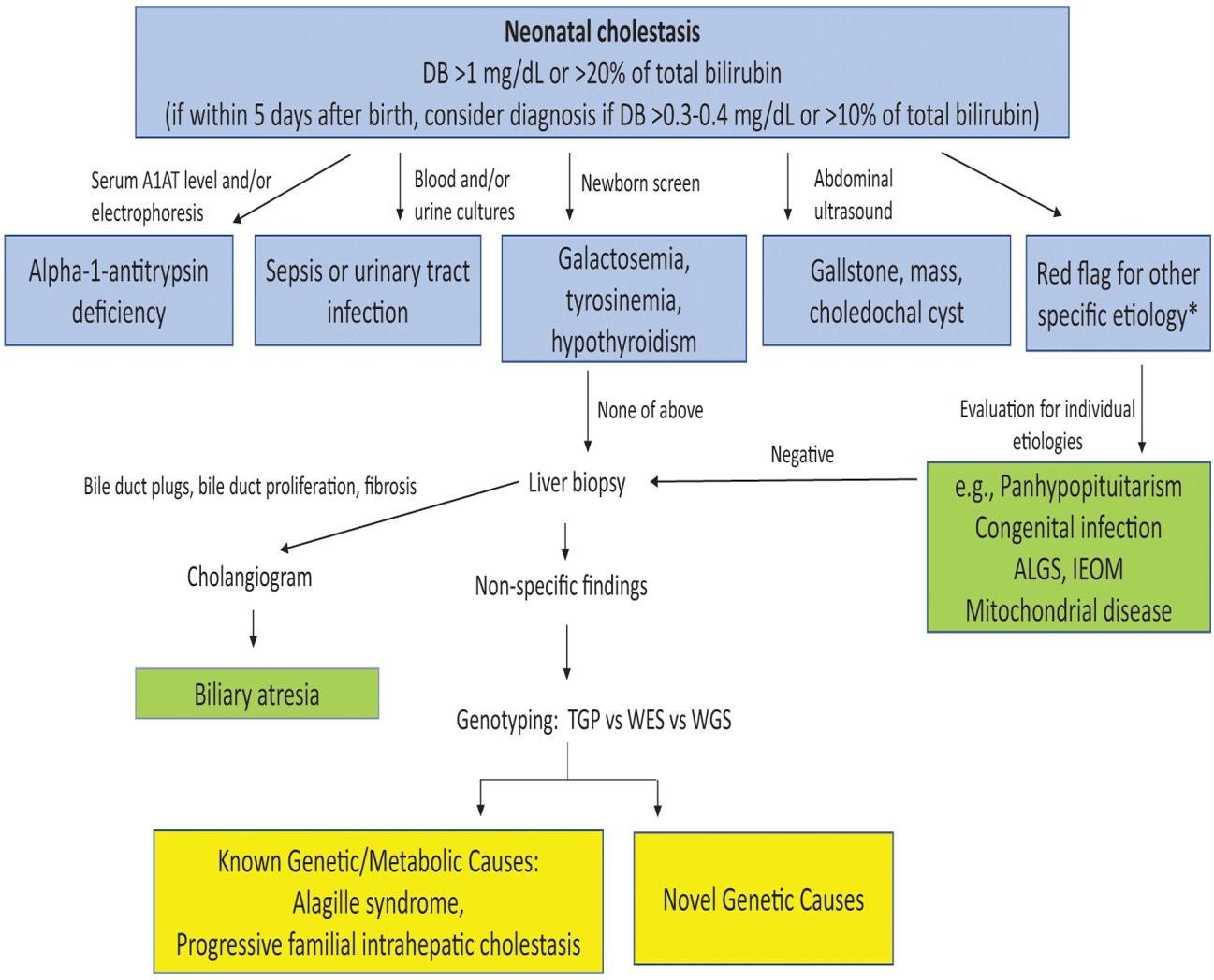
Figure 5:Algorithm for evaluation of neonatal cholestasis.
(BA, A1AT, and treatable causes of cholestasis (choledochal cyst, infection, galactosemia, tyrosinemia, hypothyroidism) are excluded in a timely fashion through blood tests, imaging, potential liver biopsy, and intraoperative cholangiography to determine if biliary atresia is present. If there is a “red flag” based on maternal history, family history, or physical examination suggesting a specific diagnosis, the infant should be promptly evaluated for that disease. If a cause is not identified, targeted gene panels, WES or WGS can be used to efficiently and effectively evaluate for both known genetic/metabolic causes of cholestasis as well as novel genetic causes [48].
Most infants with NC are underweight and need nutritional support. The goal is to provide adequate calories to compensate for steatorrhea and to prevent or treat malnutrition. The calorie requirement is approximately 125% of the recommended dietary allowance (RDA) based on ideal body weight [49]. In breastfed infants, breastfeeding should be encouraged and medium chain triglyceride (MCT) oil should be administered in a dose of 1-2 mL/kg/d in 2-4 divided doses in expressed breast milk. In older infants, a milk cereal mix fortified with MCT is preferred. Adding puffed rice powder and MCT to milk can make feeds energy dense. Essential fatty acids should constitute 2- 3% of the energy provided. Vegetable protein at 2-3 g/kg/ d is recommended [50].
According to the updated guidelines of Indian Academy of Pediatrics (IAP) 2022 for treatment of neonatal cholestasis, the following table 2 summarizes the supportive treatment of neonatal cholestasis in the form of administration of fat- soluble vitamins, calcium, magnesium and trace elements and drugs used for treatment of pruritis.
| Drug supplementation | Dose | Side effects/ comments |
|---|---|---|
| Vitamin K | 2.5 mg twice/week to 5 mg/ day oral Or IV/IM: 2–5 mg/month | None (To be given if coagulopathy is present or till conjugated jaundice is existing) |
| Vitamin D (cholecalciferol) | Oral 2,500–4,000 IU/day Or One sachet (60,000 IU) per month | Hypercalcemia, nephrocalcinosis (To be given if coagulopathy is present or till conjugated jaundice is existing). May monitor vitamin D levels every 3 monthly and treat accordingly). |
| Vitamin A | Oral 5,000–25,000 IU/day | Hepatotoxicity, hypercalcemia Pseudotumor cerebri if higher dose is used. Avoid hypervitaminosis as it can enhance liver fibrosis |
| Vitamin E | Oral 50–400 IU/day | Potentiation of vitamin K deficiency coagulopathy, diarrhea |
| Water-soluble vitamins | Twice the recommended daily allowances | None |
| Calcium supplementation | Oral 25–100 mg/kg/day | |
| Medium chain triglycerides (MCT) | 15–20% of calories | Oral supplementation in the form of oil/ powder/formula feeds containing desired MCT content |
| Pruritus (oral drugs) | ||
| Ursodeoxycholic acid | 10–20 mg/kg/day | Diarrhea, hepatotoxicity |
| Rifampicin | 10 mg/kg/day | Hepatotoxicity, drug interactions |
| Phenobarbitone | 3–10 mg/kg/day | Sedative effect, behavioral changes |
| Cholestyramine | 0.25–0.5 g/kg/day | Constipation,steatorrhea, hyperchloremic metabolic acidosis |
Table 2:General supportive treatment of neonatal cholestasis. [51].
The specific therapy and different types of investigations either screening or confirmatory for different aetiologies of neonatal cholestasis are mentioned below in table 3.
| Disease Entity | Screening | Confirmatory Investigation | Specific Treatment |
|---|---|---|---|
| Biliary atresia (BA) | Pale colored stool, high GGT |
USG: GB rudimentary/poor contractility, triangular cord sign+; liver biopsy features of BA. Peroperative cholangiogram |
Surgical: Portoenterostomy in the large majority of cases Or Hepaticojejunostomy in a few with patent confluence at porta |
| Galactosemia | Urine for non-glucose reducing substances + | Galactose-1-phosphate uridyltransferase (GAL1-PUT) | Lactose-free diet |
| Tyrosinemia | High AFP | Urinary succinyl acetone present (sample should be quickly analyzed being highly volatile substance) |
Drug treatment with NTBC [2-(2-nitro-4-trifluoromethyl benzoyl)-1,3-cyclohexanedione] and restricted
tyrosine and phenylalanine diet. Alternatively liver transplantation |
| Gestational alloimmune liver disease (earlier named as neonatal hemochromatosis) | High serum ferritin median 2,448 (415–100,000) μg/L |
Lip biopsy (submucosal gland): Iron deposition on staining Or MRI pancreas for iron deposition |
High dose of intravenous immunoglobulin ± exchange blood transfusions. If no response, then liver transplantation |
| Bile acid transporter defects (PFIC types I–VI; types I–III being common) | GGT normal/low or high; liver biopsy features | Genetic evaluation (clinical exome sequencing) |
Medical treatment for pruritus. May need surgical biliary diversion to control pruritus. Liver transplantation, if indicated |
| Alagille syndrome | High GGT; high triglycerides; eye, cardiac, and skeletal defects; paucity of bile ducts on liver biopsy | Genetic evaluation JAG1 or NOTCH2 mutation | Same as that of bile acid transporter defects with some exceptions in managing disease complications |
Table 3:Specific therapy and investigations for different types of neonatal cholestasis. [51].
Liver transplantation
It is considered as the last option or choice for treatment of NC when end stage liver disease occurs (massive fibrosis and lost synthetic liver functions) or after failure of medical conservative manageme[52].
Potential Future Therapeutics
New Advances in our understanding of bile formation and secretion, bile acid signaling, and pathogenesis of many cholestatic disorders have spurred a great deal of interest in the development of new therapies which include:
For infants with BA and other fibroinflammatory diseases, antifibrotic and anti-inflammatory medications may be of theoretical benefit, unfortunately, no benefit was demonstrated with initial trials of high dose corticosteroid or intravenous immunoglobulin for infants with BA [53].
Screening and Prevention
Early diagnosis of NC is considered of great importance to prevent liver cell fibrosis, damage and subsequent mortality especially in cases with BA which should be diagnosed as early as possible. In Taiwan, Japan, and Switzerland, a national stool color screening system has been implemented in which parents are given an infant stool color card at birth. They are instructed to notify care providers if their infant passes an acholic stool and to bring the stool color card to the 1-month well child visit to review with the provider. This program has significantly reduced the average age at diagnosis and time of HPE. In addition, it has improved the 3-month post-HPE jaundice-free rate and the 5-year overall survival rate [54].
Neonatal cholestasis can affect all body systems. Although, it is multifactorial, biliary atresia remains the most common cause. Other causes include genetic, metabolic, infections, drug induced, immune mediated and TPN associated. Clinically, presented with cholestatic jaundice, hepatosplenomegaly, ascites, purpura or bleeding manifestations, cardiac manifestation (murmur of pulmonary artery stenosis as in Alagille syndrome). Full history taking and physical examination is mandatory to discover key points that help in diagnosis. Recently, new modalities of investigations can be used in diagnosis such as ERCP, MRCP, liver biopsy and genetic testing (WES). Treatable causes of NC should be managed early to prevent long term complications.
NC: Neonatal cholestasis; NASPGHAN: North American Society of Pediatric Gastroenterology, Hepatology and Nutrition; ESPGHAN: European Society of Pediatric Gastroenterology, Hepatology and Nutrition; BA: Biliary atresia; DBL: Direct bilirubin level; ALP: Alkaline phosphatase; GGT: Gamma glutamyle transferase; CT: Computerized tomography; ERCP: Endoscopic retrograde pancreatography; A1AT: Alpha 1 antitrypsin; ALGS: Alagille syndrome; CF: Cystic fibrosis; PFIC: Progressive familial intrahepatic cholestasis; INH: Idiopathic neonatal hepatitis; TNC: Transient neonatal cholestasis; PNAC: Parenteral nutrition associated cholestasis; CBD: Common bile duct; TJs: Tight junctions; ABC: ATP binding cassette; BSEP: Bile salt export pump; BADF: Bile acid dependent fraction; GSH: Glutathione, sulphate, hyaluronic acid; MRP2: Multi drug resistance protein 2; BAIF: Bile acid independent fraction; LCHAD: Long chain hydroxy acyl- dehydrogenase; GALD: Gestational allo-immune liver disease; JAGGED: JAG1 gene protein of Alagille syndrome; NGS: Next generation sequencing; WES: Whole exon sequencing; WGS: Whole genome sequencing; HIDA: Hepatobiliary iminodiacetic acid analogue; PN: Parenteral nutrition; IOC: Intra-operative cholangiography; HPE: Hepatobiliary endoscopy; IBS: Inspissated bile syndrome; EHBD Extrahepatic bile duct; UDCA: Ursodeoxycholic acid; SPBD: Spontaneous perforation of bile duct; PBM: Pancreaticobiliary malunion; TNFἀ: Tumor necrosis factor alpha; IL1ᵝ: Interleukin 1 beta; CFTR: Cystic fibrosis transmembrane regulator; AQP1: Aquaporin channel protein 1; TAT: Tyrosine aminotransferase; FAH: Fumaryl acetoacetate hydrolase; HPD: Hydroxyoxyphenyle dioxygenase; AD: Autosomal dominant; AASLD: American Association for Society of Liver Diseases; FTT: Failure to thrive; TPN-AC: Total parenteral nutrition associated cholestasis; RDA: Recommended daily allowance; MCT: Medium chain triglycerides; AFP: Alpha fetoprotein; NTBC: Nitro-trifluoromethylphenyl cyclohexanedione; IAP: Indian academy of Pediatrics; MMP7: Matrix metalloproteinase 7; FXR: Farensoid X receptor
Early identification and diagnosis of NC is mandatory especially for BA, which needs early surgical intervention to improve morbidity and mortality. Many screening tools are recommended such as acholic stool monitoring, mobile health application and MMP-7 measurement which is increased in cases with BA. In addition to the ordinary neonatal screening programs that can diagnose early genetic, metabolic causes of NC which prevent progression to liver cell failure. It is also recommended to refer a case of cholestasis to a specialized hospital when diagnosed to be under supervision of multidisciplinary team.